Calculation of electronic absorption spectra#
This section discusses the calculation of electronic absorption spectra using the real time propagation of electronic dynamics as implemented in DFTB+.
Unless some good reason exists for not doing so, the electronic spectrum should be calculated at the equilibrium geometry. For this example we will use an optimised chlorophyll a molecule. This example reproduces the results in Oviedo, M. B., Negre, C. F. A., & Sánchez, C. G. (2010). Dynamical simulation of the optical response of photosynthetic pigments. Physical Chemistry Chemical Physics : PCCP, 12(25), 6706–6711.
The input#
[Input: recipes/electronicdynamics/spectrum/]
The following input can be used to calculate the absorption spectrum of chlorophyll a:
InputVersion = 20.1
Geometry = GenFormat {
<<< "coords.gen"
}
Driver {}
Hamiltonian = DFTB {
Scc = Yes
SccTolerance = 1.0e-7
SlaterKosterFiles = Type2FileNames {
Prefix = "../../slakos/download/3ob/3ob-3-1/"
Separator = "-"
Suffix = ".skf"
}
MaxAngularMomentum {
Mg = "p"
C = "p"
N = "p"
O = "p"
H = "s"
}
Filling = Fermi {
Temperature [K] = 300
}
}
ElectronDynamics {
Steps = 20000
TimeStep [au] = 0.2
Perturbation = Kick {
PolarizationDirection = "all"
}
FieldStrength [V/A] = 0.001
}
The optimised geometry is located in the coords.gen file. Note that for this example the long phytol chain present in the natural molecule has been replaced by a terminating hydrogen atom since it does not have a significant influence on the absorption spectrum.
For the calculation of absorption spectra, an initial kick of the system is made
using a Dirac delta type perturbation. The input specifies that after the
initial perturbation of Kick type, twenty thousand steps of dynamics will be
executed using a time step of 0.2 atomic units. The Kick perturbation can be
applied in any of the Cartesian directions (x, y or z). The use of
all here in the input instructs the code to run three independent dynamic
calculations, one with an initial Kick in each Cartesian direction.
After self consistency has been achieved and the ground state density matrix is obtained, the perturbation is applied and then the propagation starts, the output produced is the following:
S inverted
Density kicked along x!
Starting dynamics
Step 0 elapsed loop time: 0.012400 average time per loop 0.012400
Step 2000 elapsed loop time: 19.112000 average time per loop 0.009551
Step 4000 elapsed loop time: 35.407101 average time per loop 0.008850
Step 6000 elapsed loop time: 52.179100 average time per loop 0.008695
Step 8000 elapsed loop time: 68.688004 average time per loop 0.008585
Step 10000 elapsed loop time: 90.615501 average time per loop 0.009061
Step 12000 elapsed loop time: 109.174500 average time per loop 0.009097
Step 14000 elapsed loop time: 127.921097 average time per loop 0.009137
Step 16000 elapsed loop time: 147.406097 average time per loop 0.009212
Step 18000 elapsed loop time: 167.002502 average time per loop 0.009277
Step 20000 elapsed loop time: 185.372406 average time per loop 0.009268
Dynamics finished OK!
S inverted
Density kicked along y!
Starting dynamics
Step 0 elapsed loop time: 0.023700 average time per loop 0.023700
Step 2000 elapsed loop time: 28.003799 average time per loop 0.013995
Step 4000 elapsed loop time: 52.257900 average time per loop 0.013061
Step 6000 elapsed loop time: 74.137497 average time per loop 0.012354
Step 8000 elapsed loop time: 93.527603 average time per loop 0.011689
Step 10000 elapsed loop time: 115.045998 average time per loop 0.011503
Step 12000 elapsed loop time: 134.955200 average time per loop 0.011245
Step 14000 elapsed loop time: 155.862000 average time per loop 0.011132
Step 16000 elapsed loop time: 176.434799 average time per loop 0.011026
Step 18000 elapsed loop time: 197.430695 average time per loop 0.010968
Step 20000 elapsed loop time: 217.860703 average time per loop 0.010892
Dynamics finished OK!
S inverted
Density kicked along z!
Starting dynamics
Step 0 elapsed loop time: 0.012100 average time per loop 0.012100
Step 2000 elapsed loop time: 27.119101 average time per loop 0.013553
Step 4000 elapsed loop time: 48.640301 average time per loop 0.012157
Step 6000 elapsed loop time: 67.843803 average time per loop 0.011305
Step 8000 elapsed loop time: 87.514702 average time per loop 0.010938
Step 10000 elapsed loop time: 111.822601 average time per loop 0.011181
Step 12000 elapsed loop time: 133.397202 average time per loop 0.011116
Step 14000 elapsed loop time: 153.044098 average time per loop 0.010931
Step 16000 elapsed loop time: 176.008301 average time per loop 0.011000
Step 18000 elapsed loop time: 195.700104 average time per loop 0.010872
Step 20000 elapsed loop time: 216.208694 average time per loop 0.010810
Dynamics finished OK!
The resulting time dependent dipole moment along each Cartesian direction produced the kicks are stored in the mu*.dat output files.
The calculation of the spectrum makes use of the fact that the Fourier transform of induced dipole moment of the molecule in the presence of an external time dependent field (within the linear response range) is related to the Fourier transform of said field in the following manner:
since the Fourier transform of a Dirac delta is a constant at all frequencies, the polarizability tensor \(\overset\leftrightarrow{\alpha}(\omega)\) can be obtained from the time dependent response. The absorption is proportional to the imaginary part of the trace of the polarizability tensor.
The calculation of the absorption spectrum is carried out using the script
calc_timeprop_spectrum either available after make install of DFTB+, or
located in the tools/misc directory under the dftbplus source tree. The
invocation of the script is as follows:
calc_timeprop_spectrum -d 20.0 -f 0.001
The exciting field strength is specified with the -f flag, the -d flag specifies a damping constant used to exponentially damp the dipole signal to zero within the simulation time. This damping time is expressed in femtoseconds. The effect of damping the dipole moment is to add a uniform width to every spectral line and is necessary to smooth out any ringing in the spectrum peaks after the transform. In essence this damping procedure is equivalent to using a windowing function.
The spectrum is located in the output files spec-ev and spec-nm. In this case the spectrum looks as follows:
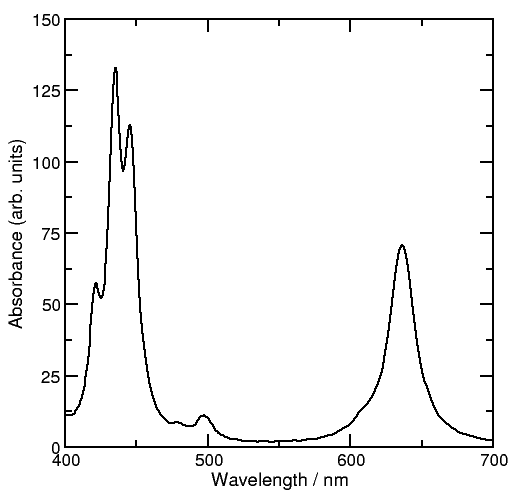
The band between 400 and 500 nm is called the Soret band and the one between 600 and 700 nm is the Q band. This band is the band that provides is responsible for the photo-biologic activity of chlorophylls as antennae capable of capturing solar energy in the primary process of photosynthesis.